Quantum Circuit Tutorial#
In the ansatz tutorial of the basic tutorial, we demonstrated how to use the Unity Coupled Cluster, Hardware-Efficient, and Symmetry-Reserved interfaces to directly build quantum circuits. This tutorial demonstrates how to use a user-defined method to build quantum circuits.
By calling the UserDefine function under the pychemiq.Circuit.Ansatz module, we can customize the construction of the quantum circuit in two ways: the first is to input the originIR format code into the circuit parameter to customize the quantum circuit, and the second is to construct the quantum circuit by inputting the fermionic operator fermion parameter of the coupled cluster excitation term. The interface of this function is introduced as follows:
- UserDefine(n_electrons, circuit=None, fermion=None, chemiq=None)#
Construct quantum circuit ansatz using user-defined methods.
- Parameters:
n_electrons (int) – Enter the number of electrons in the molecular system.
circuit (str) – Construct the originIR string of a quantum circuit.
fermion (FermionOperator) – The fermionic operator class for constructing quantum circuits.
chemiq (ChemiQ) – Specify the chemiq class. Please refer to pychemiq.ChemiQ for details.
- Returns:
Outputs the customized ansatz AbstractAnsatz class.
1. Customize the quantum circuit by inputting originIR into the circuit parameter
We can obtain quantum circuits in originIR format through two methods. The first method is to build quantum circuits through the graphical programming interface of the origin quantum cloud platform. As shown in the following figure, first build a quantum circuit on the original quantum cloud platform Graphical programming interface.
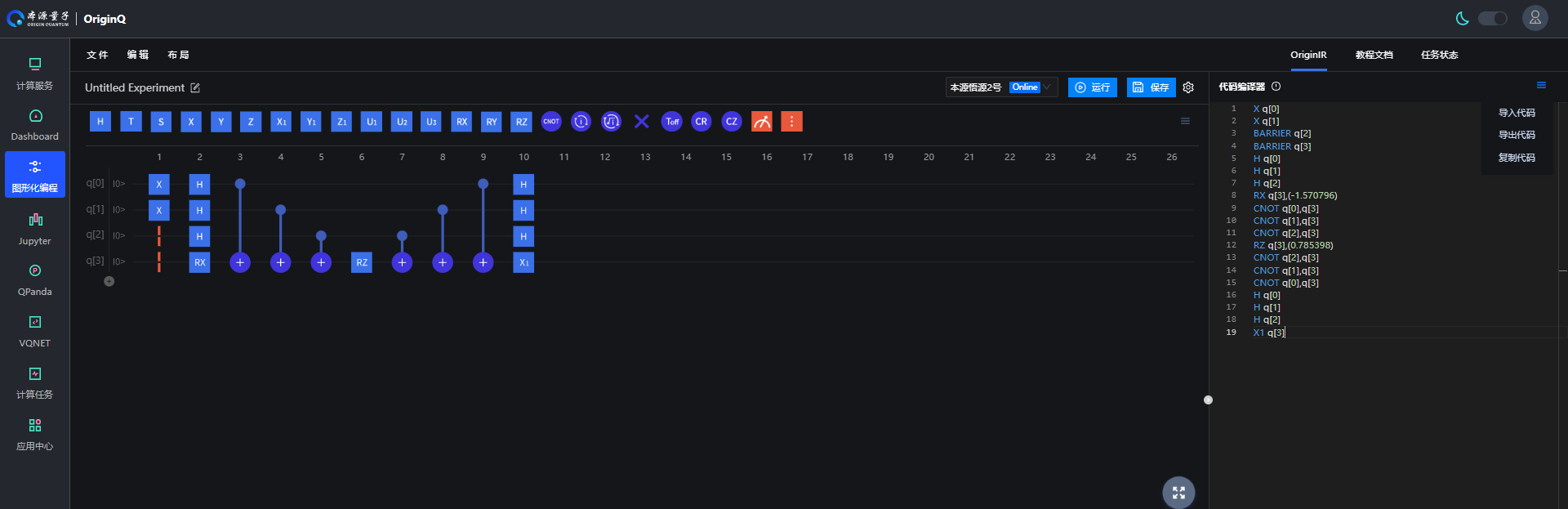
Figure 1: Building quantum circuits through the graphical programming interface of the origin quantum cloud platform
After building the quantum circuit, export the following originIR format string and input it into the circuit parameter in the UserDefine function.
X q[0]
X q[1]
BARRIER q[2]
BARRIER q[3]
H q[0]
H q[1]
H q[2]
RX q[3],(-1.570796)
CNOT q[0],q[3]
CNOT q[1],q[3]
CNOT q[2],q[3]
RZ q[3],(0.785398)
CNOT q[2],q[3]
CNOT q[1],q[3]
CNOT q[0],q[3]
H q[0]
H q[1]
H q[2]
X1 q[3]
Interface Example:
from pychemiq import Molecules,ChemiQ,QMachineType
from pychemiq.Transform.Mapping import jordan_wigner,MappingType
from pychemiq.Optimizer import vqe_solver
from pychemiq.Circuit.Ansatz import UserDefine
import numpy as np
multiplicity = 1
charge = 0
basis = "sto-3g"
geom = "H 0 0 0,H 0 0 0.74"
mol = Molecules(
geometry = geom,
basis = basis,
multiplicity = multiplicity,
charge = charge)
fermion_H2 = mol.get_molecular_hamiltonian()
pauli_H2 = jordan_wigner(fermion_H2)
chemiq = ChemiQ()
machine_type = QMachineType.CPU_SINGLE_THREAD
mapping_type = MappingType.Jordan_Wigner
pauli_size = len(pauli_H2.data())
n_qubits = mol.n_qubits
n_elec = mol.n_electrons
chemiq.prepare_vqe(machine_type,mapping_type,n_elec,pauli_size,n_qubits)
# Using a custom quantum circuit, input the originIR format string into the circuit parameter
circuit = """
X q[0]
X q[1]
BARRIER q[2]
BARRIER q[3]
H q[0]
H q[1]
H q[2]
RX q[3],(-1.5707963)
CNOT q[0],q[3]
CNOT q[1],q[3]
CNOT q[2],q[3]
RZ q[3],(0.785398)
CNOT q[2],q[3]
CNOT q[1],q[3]
CNOT q[0],q[3]
H q[0]
H q[1]
H q[2]
RX q[3],(1.5707963)
"""
ansatz = UserDefine(n_elec, circuit=circuit, chemiq=chemiq)
# Finally, specify the classical optimizer and initial parameters and iteratively solve them
method = "SLSQP"
init_para = np.zeros(ansatz.get_para_num())
solver = vqe_solver(
method = method,
pauli = pauli_H2,
chemiq = chemiq,
ansatz = ansatz,
init_para=init_para)
result = solver.fun_val
print(result)
The printed result is: 0.7151043390810803。 The two RX gates here are fixed parameters and do not participate in parameter optimization of variational circuits. For revolving doors with parameters, the default parameters that are not \(\pi /2\) or \(- \pi /2\) are the parameters to be optimized.
The second way to obtain the originIR format of quantum circuits is through the convert_qprog_to_originir function in pyqpanda, as detailed in Quantum Program Conversion OriginIR . Here, we take the Hardware-Efficient single-layer circuit ansatz in the ansatz tutorial as an example to demonstrate how to obtain OriginIR through quantum programming. Next, we will first construct the HE ansatz line QProg, and then convert it into originIR format through the function convert_qprog_to_originir(prog, machine).
import pyqpanda as pq
import numpy as np
def HE_ansatz(machine_type,qn, para):
machine = pq.init_quantum_machine(machine_type)
qlist=pq.qAlloc_many(qn)
# Build HE ansatz line
prog = pq.QProg()
for i in range(qn):
prog.insert(pq.RZ(qlist[i], para[4*i]))
prog.insert(pq.RX(qlist[i], para[4*i+1]))
prog.insert(pq.RZ(qlist[i], para[4*i+2]))
for j in range(qn-1):
ry_control = pq.RY(qlist[j+1], para[4*j+3]).control(qlist[j])
prog.insert(ry_control)
ry_last = pq.RY(qlist[0], para[4*qn-1]).control(qlist[qn-1])
prog.insert(ry_last)
#print(prog)
OriginIR=pq.convert_qprog_to_originir(prog, machine)
print(OriginIR)
return OriginIR
Below, we define the main function to obtain the originIR format quantum circuit under this parameter. Here we take four qubits as examples:
if __name__ == "__main__":
machine_type = pq.QMachineType.CPU
qn=4
para=np.random.random(4*qn)
HE_ansatz(machine_type,qn, para)
The printed result is:
QINIT 4
CREG 0
RZ q[0],(0.6639123)
RX q[0],(0.69876429)
RZ q[0],(0.87923246)
RZ q[1],(0.50633782)
RX q[1],(0.57366393)
RZ q[1],(0.51500428)
RZ q[2],(0.41510053)
RX q[2],(0.58136057)
RZ q[2],(0.60506401)
RZ q[3],(0.99153126)
RX q[3],(0.89568316)
RZ q[3],(0.6493124)
CONTROL q[0]
RY q[1],(0.011800026)
ENDCONTROL
CONTROL q[1]
RY q[2],(0.92157183)
ENDCONTROL
CONTROL q[2]
RY q[3],(0.64791654)
ENDCONTROL
CONTROL q[3]
RY q[0],(0.50756615)
ENDCONTROL
After deleting the first two lines, you can input them into the circuit parameter in the UserDefine function, as shown in the first method.
2. Construct quantum circuit by inputing “fermion” parameters, which are fermionic operators obtained from coupled cluster operators
The second method isconstruct quantum circuit by inputing “fermion” parameters, which are fermionic operators obtained from coupled cluster operators. For example, for 4-qubits, the double excited coupling cluster operator of a 2-electron system has spin orbitals 0 and 1 as occupied states, and the excited coupling cluster term is: 01->23.

Figure 2: Hydrogen molecular system with four spin orbitals from ground state to double excited state
To construct the excitation fermionic operator mentioned above, we need to use the FermionOperator or call the function get_cc() in the pychemiq.Utils module to construct it.
from pychemiq import FermionOperator
a = FermionOperator("3+ 2+ 1 0", 1)
print(a)
from pychemiq.Utils import get_cc_n_term,get_cc
import numpy as np
n_para = get_cc_n_term(4,2,"CCD")
para = np.ones(n_para)
cc_fermion = get_cc(4,2,para,"CCD")
print(cc_fermion)
The printed results of both are:
{
3+ 2+ 1 0 : 1.000000
}
Input the obtained excitation fermionic operators as the ‘fermion’ parameter into the ‘UserDefine’ function. Here, we use the example of a hydrogen molecule:
Interface example:
from pychemiq import Molecules,ChemiQ,QMachineType,FermionOperator
from pychemiq.Transform.Mapping import jordan_wigner,MappingType
from pychemiq.Optimizer import vqe_solver
from pychemiq.Circuit.Ansatz import UserDefine
import numpy as np
multiplicity = 1
charge = 0
basis = "sto-3g"
geom = "H 0 0 0,H 0 0 0.74"
mol = Molecules(
geometry = geom,
basis = basis,
multiplicity = multiplicity,
charge = charge)
fermion_H2 = mol.get_molecular_hamiltonian()
pauli_H2 = jordan_wigner(fermion_H2)
chemiq = ChemiQ()
machine_type = QMachineType.CPU_SINGLE_THREAD
mapping_type = MappingType.Jordan_Wigner
pauli_size = len(pauli_H2.data())
n_qubits = mol.n_qubits
n_elec = mol.n_electrons
chemiq.prepare_vqe(machine_type,mapping_type,n_elec,pauli_size,n_qubits)
# Using a custom quantum circuit, input the custom excitation fermionic operator into the fermion parameter
a = FermionOperator("3+ 2+ 1 0", 1)
ansatz = UserDefine(n_elec, fermion=a, chemiq=chemiq)
# Finally, specify the classical optimizer and initial parameters and iteratively solve them
method = "SLSQP"
init_para = np.zeros(ansatz.get_para_num())
solver = vqe_solver(
method = method,
pauli = pauli_H2,
chemiq = chemiq,
ansatz = ansatz,
init_para=init_para)
result = solver.fun_val
print(result)
The printed result is: -1.1372838304374302