Algorithm Tutorial#
Since the conception of quantum computing was introduced, hundreds of quantum algorithms have been developed [1] . Although there is significant interest in simulating chemical problems on quantum computers, only a small number of quantum algorithms currently serve as the foundation for designing such quantum programs. Among the quantum algorithms applied to material simulation and biomedical problems, many require substantial quantum resources, such as a large number of qubits or deep quantum circuits. In contrast, the Variational Quantum Eigensolver, a hybrid quantum-classical algorithm, leverages the computational power of noisy qubits to approximately solve relevant problems without the need for full error correction.
The Variational Quantum Eigensolver (VQE) algorithm [2] is a hybrid quantum-classical algorithm that uses parameterized quantum circuits to construct wave functions and optimizes these parameters via a classical computer to minimize the expectation value of the Hamiltonian. As shown in Figure 1, the overall workflow includes quantum initial state preparation, measurement of Hamiltonian sub-terms, summation of all the sub-terms, convergence judgment, and parameter optimization. Among them, quantum state preparation and measurement of sub-terms of Hamiltonian (also known as quantum expectation estimation) is carried out on the quantum computer, that is, the light yellow part in the figure, and other steps such as summation and parameter optimization are carried out by the classical computer , which is the light blue part in the figure.
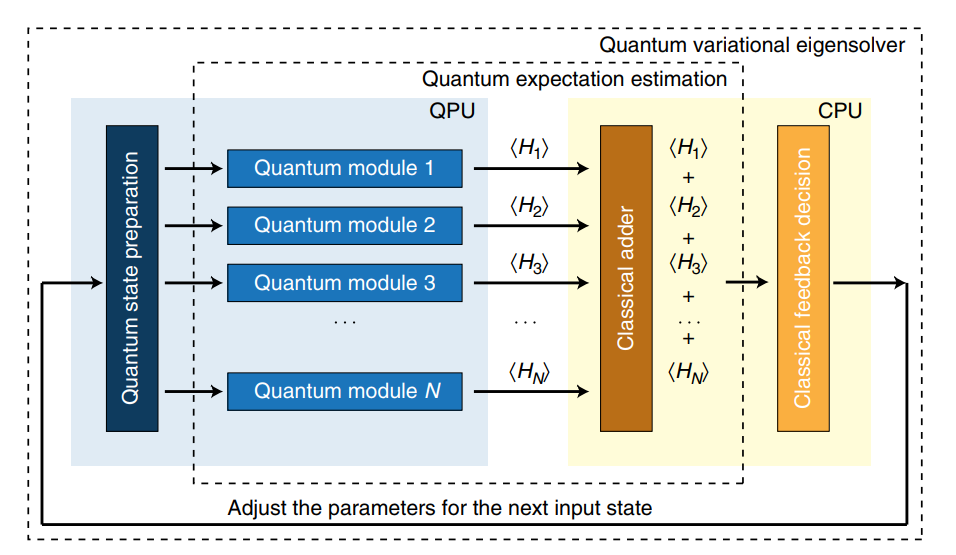
Figure 1: VQE Algorithm Process
Specifically, VQE can be summarized as the following steps:
Select a set of random initial parameters :math:` theta_1^k, theta_2^k, theta_3^k ldots theta_n^k`.
Prepare a trial wave function \(|\psi(\vec{\theta})\rangle\) in the virtual machine or a quantum computer.
Measure each component of the Hamiltonian, and then sum them up on a classical computer to get the expected value of the Hamiltonian for the trial wave function \(|\psi(\vec{\theta})\rangle\), that is, the energy of the molecule.
Judge whether the energy satisfies the convergence criteria, if so, use the value as the approximate energy of the molecule ground state energy and terminate the calculation; if not, then the parameters got optimized in a variational way to generate a new set of parameters \(\theta_1^{k+1}, \theta_2^{k+1}, \theta_3^{k+1} \ldots \theta_n^{k+1}\) using a classical optimizer and prepare a new quantum state.
Repeat (ii)-(iv) until the energy converges.
At this point, the parameterized quantum circuit has prepared the ground state of the Hamiltonian, or a state very close to the ground state. Comparing to quantum phase estimation algorithm, VQE requires fewer gates and shorter coherence times. VQE exchanges the long coherence times needed for phase estimation for a polynomial overhead due to measurement repetitions and classical processing. Therefore, it is more suitable for the NISQ era.
In the entire algorithm, the first step of quantum initial state preparation is crucial for quickly obtaining correct results, especially when applied to chemical systems, as electrons are fermions, some additional preprocessing is essential. The quantum initial state is typically the Hartree-Fock state, which is obtained through classical computations. However, different mapping methods also influence the construction of the initial state. Commonly used mapping methods include the Jordan-Wigner (JW) transformation, Parity transformation, and Bravyi-Kitaev (BK) transformation. After determining the mapping method and construction the initial state for the circuit, we need a suitable wavefunction ansatz to obtain a trial wavefunction \(|\psi (\theta) \rangle\) that approximates the system’s final quantum state.
At present, the preparation methods of trial states in quantum computing chemistry are mainly divided into two categories. One is traditional computational chemistry inspired ansatzes, such as the Unitary Coupled Cluster (UCC) method, and the other is ansatzes constructed based on the hardware topology of quantum computers, namely Hardware-Efficient ansatzes. Once a ansztz is selected, its corresponding circuit can be executed on a quantum computer to calculate the objective function value, and the corresponding result in VQE is the expected energy. After multiple iterations of measurement until convergence, the final state obtained is the quantum state closest to the true system ground-state, and the corresponding energy is the obtained ground-state energy.
In the case calculation of \(H_2O\) molecules below, we use the sto-3g basis set, active space [4,4], mapping method using BK transformation, ansatz using UCCSD. The classical optimizer method uses the first-order derivative optimization method L-BFGS-B:
from pychemiq import Molecules,ChemiQ,QMachineType
from pychemiq.Transform.Mapping import bravyi_kitaev,MappingType
from pychemiq.Optimizer import vqe_solver
from pychemiq.Circuit.Ansatz import UCC
import numpy as np
# Initialize the electronic structure parameters of molecules, including charge, base group, atomic coordinates, spin multiplicity, and active space
multiplicity = 1
charge = 0
basis = "sto-3g"
geom = ["O 0.00000000 0.00000000 0.12713100",
"H 0.00000000 0.75801600 -0.50852400",
"H 0.00000000 -0.75801600 -0.50852400"]
active = [4,4]
mol = Molecules(
geometry = geom,
basis = basis,
multiplicity = multiplicity,
charge = charge,
active = active)
# Obtain the Hamiltonian of water molecules in the form of Pauli operators through BK transformation and print the results
fermion_H2O = mol.get_molecular_hamiltonian()
pauli_H2O = bravyi_kitaev(fermion_H2O)
print(pauli_H2O)
#To prepare a quantum circuit, the parameters that need to be specified include the quantum virtual machine type machine_type, intended mapping_type,
#The number of terms pauli_size, the number of electrons n_elec, and the number of qubits n_qubits of the Pauli Hamiltonian
chemiq = ChemiQ()
machine_type = QMachineType.CPU_SINGLE_THREAD
mapping_type = MappingType.Bravyi_Kitaev
pauli_size = len(pauli_H2O.data())
n_qubits = mol.n_qubits
n_elec = mol.n_electrons
chemiq.prepare_vqe(machine_type,mapping_type,n_elec,pauli_size,n_qubits)
# The mapping method and type of cluster operator required for setting cluster operators are ansatzed using UCCSD
ansatz = UCC("UCCSD",n_elec,mapping_type,chemiq=chemiq)
# Specify classic optimizer and initial parameters and iteratively solve
method = "L-BFGS-B"
init_para = np.zeros(ansatz.get_para_num())
solver = vqe_solver(
method = method,
pauli = pauli_H2O,
chemiq = chemiq,
ansatz = ansatz,
init_para=init_para)
result = solver.fun_val
n_calls = solver.fcalls
print(result,f"function called {n_calls} times")
energies = chemiq.get_energy_history()
print(energies)
The results obtained are as follows:
-74.97462360159876 function called 16 times
[-74.96590114589256, -74.93763769775363, -74.97445942068707, -74.97445942068707, -74.97411682452937, -74.9746226763453, -74.9746226763453, -74.97462062772358, -74.97462337673937, -74.97462337673937, -74.97462142026288, -74.97462351765488, -74.97462351765488, -74.974622639902, -74.97462360159876, -74.97462360159876]
In order to compare the computational accuracy of pyChemiq, we compared the results with the results of the classic computational chemistry software PySCF [3] (see installation details for PySCF website). In PySCF, we used the same basis set and method (UCCSD ansatz in VQE corresponds to the classic CISD method), with the following code:
from pyscf import gto, scf
atom = '''
O 0.00000000 0.00000000 0.12713100
H 0.00000000 0.75801600 -0.50852400
H 0.00000000 -0.75801600 -0.50852400
'''
mol = gto.M(atom=atom, # in Angstrom
basis='STO-3G',
charge=0,
spin=0)
myhf = mol.RHF().run()
mycas = myhf.CASCI(4, 4).run()
E_CISD = mycas.e_tot
print(E_CISD)
The results obtained are as follows:
converged SCF energy = -74.9659011458929
CASCI E = -74.9746354406465 E(CI) = -6.11656024435146 S^2 = 0.0000000
-74.9746354406465
We will plot the data printed by pyChemiQ and compare it with classic CISD results at the same level. It can be seen that as the number of iterations of the function increases, the electron energy gradually converges to the energy of the classical result, as shown in Figure 2. And by the fifth iteration of the function, the electron energy had already reached chemical accuracy \(1.6\times 10^{-3}\) Hartree.
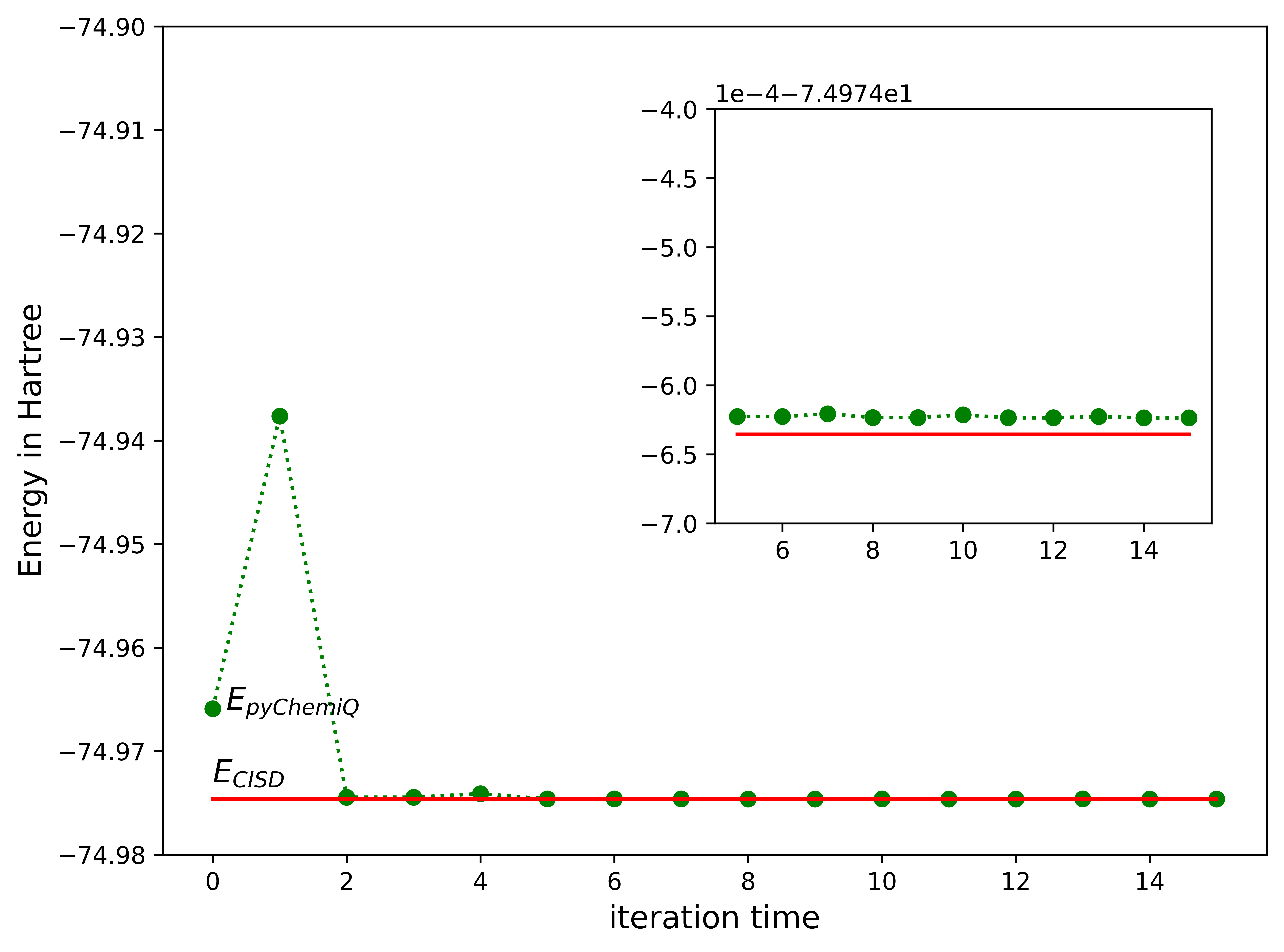
Figure 2: Energy Convergence Curve of Water Molecules
References