Mapping Tutorial#
In order to simulate electronic structure problems on quantum computers, we need a mapping relationship that maps the fermionic operator of the electron to the Pauli operator (i.e. Pauli X matrix, Pauli Y matrix, Pauli Z matrix, identity matrix I) on the quantum computer. Currently,common mappings include the Jordan-Wigner (JW) transform [1] Bravyi-Kitaev (BK) transformation [2] and the Parity transformation [3] etc. While different transformations may result in various quantum circuit depths, their functionalities are consistent — all aiming to map the fermionic system onto a quantum computer.
Here, we use the JW transformation as an example to display the mapping diagrams of three spin orbitals, as shown in Figure 1. It can be seen that under the JW transformation, each qubit identifies a fermionic orbital, and the occupied and unoccupied states are mapped to the: \(|1\rangle\) state and \(|0\rangle\) state of the qubits, respectively. At this point, the orbitals and qubits correspond one-to-one.
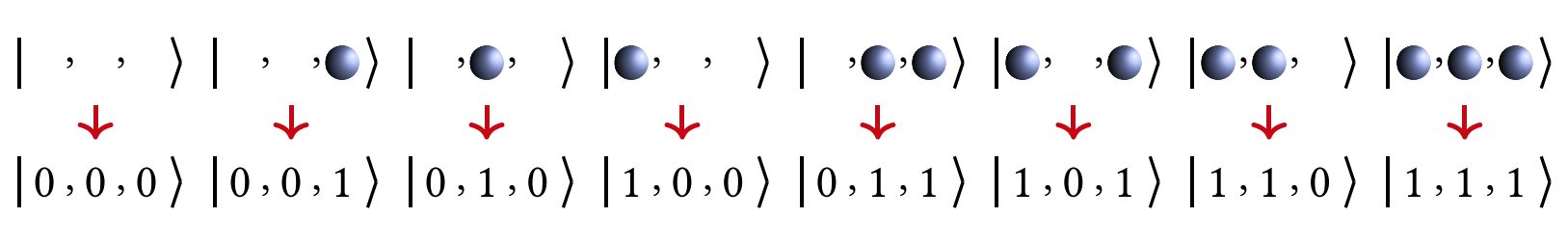
Figure 1: Schematic diagram of the JW transformation of three spin orbitals. The figure is cited from [4]
In the pychemiq. Transform module, a very important submodule is pychemiq. Transform. Mapping, which maps fermionic operators to Pauli operators. The current mapping methods supported by pyChemiQ include Jordan-Wigner (JW) transformation, Bravyi-Kitaev (BK) transformation, Parity transformation, and Multilayer Segmented transformation
Parity (MSP) transformation [5]. You can invoke the corresponding package in the following way:
from pychemiq.Transform.Mapping import (
jordan_wigner,
bravyi_kitaev,
parity,
segment_parity)
For example, using the JW transformation, we can map the fermionic Hamiltonian of the hydrogen molecule from the previous section into its Pauli form. The example code and printed results are as follows:
# First initialize to obtain the Fermion Hamiltonian of the hydrogen molecule
from pychemiq import Molecules
multiplicity = 1
charge = 0
basis = "sto-3g"
geom = "H 0 0 0,H 0 0 0.74"
mol = Molecules(
geometry = geom,
basis = basis,
multiplicity = multiplicity,
charge = charge)
fermion_H2 = mol.get_molecular_hamiltonian()
# Mapping the Fermion Hamiltonian of the obtained hydrogen molecule into Pauli form using JW transformation
pauli_H2 = jordan_wigner(fermion_H2)
print(pauli_H2)
{
"" : -0.097066,
"X0 X1 Y2 Y3" : -0.045303,
"X0 Y1 Y2 X3" : 0.045303,
"Y0 X1 X2 Y3" : 0.045303,
"Y0 Y1 X2 X3" : -0.045303,
"Z0" : 0.171413,
"Z0 Z1" : 0.168689,
"Z0 Z2" : 0.120625,
"Z0 Z3" : 0.165928,
"Z1" : 0.171413,
"Z1 Z2" : 0.165928,
"Z1 Z3" : 0.120625,
"Z2" : -0.223432,
"Z2 Z3" : 0.174413,
"Z3" : -0.223432
}
In addition, pyChemiQ also supports self constructing fermionic operators or Pauli operators to customize Hamiltonians. Please refer to the first section of the advanced tutorial for details.
References