Cases of Hydrogen Molecular Computing (Hydrogen Molecule as Case Study)#
To quickly get started with pyChemiQ, let’s first take a look at the case calculation of \(H_2\). Before setting parameters, let’s first introduce two nouns: mapping and ansatz. To simulate electronic structure problems on a quantum computer, we require a set of transformation methods to encode the fermionic operators of electrons into the Pauli operators of the quantum computer. This process is referred to as mapping. To obtain a trial wavefunction close to the true ground state of the quantum system, we need an appropriate wavefunction assumption. In principle, the closer the assumed trial state is to the ideal wavefunction, the more favorable it is for accurately obtaining the ground-state energy later on.
In the following example of hydrogen molecules, we use the STO-3G basis set, the Jordan-Wigner (JW) transformation for mapping, the UCCSD ansatz, and the SLSQP classical optimizer. We will not dive into the theoretical background behind each method here now. For a detailed introduction, please refer to the theoretical background section:
# Firstly import the required packages
from pychemiq import Molecules,ChemiQ,QMachineType
from pychemiq.Transform.Mapping import jordan_wigner,MappingType
from pychemiq.Optimizer import vqe_solver
from pychemiq.Circuit.Ansatz import UCC
import numpy as np
# Initialize the electronic structure parameters of molecules, including charge, basis-set, atomic coordinates (angstrom), spin multiplicity
multiplicity = 1
charge = 0
basis = "sto-3g"
geom = "H 0 0 0,H 0 0 0.74"
mol = Molecules(
geometry = geom,
basis = basis,
multiplicity = multiplicity,
charge = charge)
# Obtaining the Hamiltonian of hydrogen molecules in the form of Pauli operators using the JW transformation
fermion_H2 = mol.get_molecular_hamiltonian()
pauli_H2 = jordan_wigner(fermion_H2)
# To prepare a Quantum circuit, the parameters that need to be specified include the quantum virtual machine type:machine_type, intended mapping type:mapping_type,
# The number of terms of Pauli Hamiltonian Size : pauli_size, number of electrons : n_elec and the Number of Quantum Bits : n_qubits
chemiq = ChemiQ()
machine_type = QMachineType.CPU_SINGLE_THREAD
mapping_type = MappingType.Jordan_Wigner
pauli_size = len(pauli_H2.data())
n_qubits = mol.n_qubits
n_elec = mol.n_electrons
chemiq.prepare_vqe(machine_type,mapping_type,n_elec,pauli_size,n_qubits)
# Set the ansatz type, here we use UCCSD Ansatz
ansatz = UCC("UCCSD",n_elec,mapping_type,chemiq=chemiq)
# Specify classic optimizer and initial parameters and solve iteratively
method = "SLSQP"
init_para = np.zeros(ansatz.get_para_num())
solver = vqe_solver(
method = method,
pauli = pauli_H2,
chemiq = chemiq,
ansatz = ansatz,
init_para=init_para)
result = solver.fun_val
n_calls = solver.fcalls
print(result,f"Function called {n_calls}times in total")
energies = chemiq.get_energy_history()
print(energies)
The printed result is:
-1.1372838317140834 Function called 9 times in total
[-1.1167593073964257, -1.0382579032966825, -1.137282968297819, -1.137282968297819, -1.1372838302540205, -1.137283647727291, -1.1372838297780967, -1.1372838317140834, -1.1372838317140834]
We ploted the data printed by pyChemiQ and compare it with the classic Full CI under the same basic set. It can be seen that as the number of iterations of the function increases, the electron energy gradually converges to the energy of Full CI, as shown in the following figure. When the function iterates to the second time, the electron energy has already reached chemical accuracy \(1.6\times 10^{-3}\) Hartree.
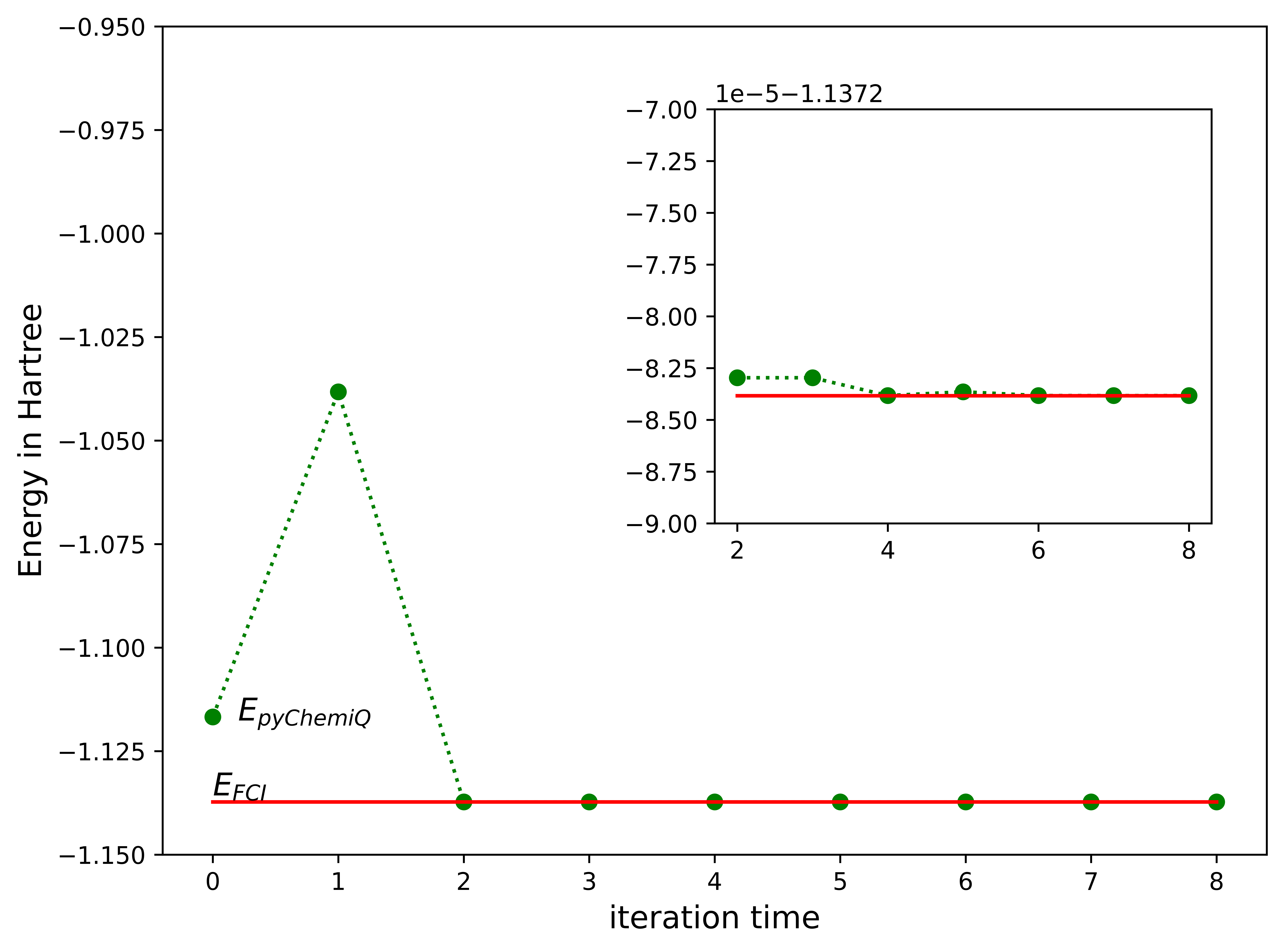
Figure : Energy Convergence Curve of Hydrogen Molecule